前言
我们已经通过ExpressionSet类和BamFileList文件(含有BAM比对后的文件)来了解了一些关于数据管理的基本知识。
- S4类有助于控制某个数据结构中不同组件之间的复杂程度和一致性。
- R包能够进一步用于协调和发布数据、软件程度和文档。在这个课程的后面部分里我们会学习如何构建R包。
在这个一部分的结尾处,我们来解决一个明显没有最佳解决方案的问题。我们考虑打包GRanges的序列化后的实例以及已经建立了索引的压缩文本文件,这些文件都可以使用GenomicFiles进行并行计算处理。
为什么这是一个比较难解决的问题呢?因为在某些情况下,含有大量功能的GRanges实例将会非常大,加载它们可能会花费大量的时间,并且会占用大量的内存。这可能导致在RAM有限时,在群集节点上使用这些表示方法出现困难。如果数据以允许随机访问的索引格式保存在磁盘上,则根据需要将文件内容的目标提取,并转换为GRanges,就可能为某些应用程度带来更好的性能。创建erma包的一个原因就是获取关于这些不同方法之间平衡后的数据。
erma:表观遗传路线图简介
为了促进对DNA突变体的理解,我们对细胞类型之间的表观基因组变异有所兴趣。NIH表观基因组路线图收集了很多关于ChIP-seq实验分析的数据,在这个公众数据库里记录了数百个不同细胞类型和标志特异性文件供其他研究者使用。
管理这类数据的有效方法是什么呢?其中erma 包就提供了这样一种方法。
BED文件集合
如同Ernst和Kellis使用大量细胞系的数据所报道的那样,他们已经进行了染色质状态的统计“估算”,如下所示:
|
|
使用GenomicFiles进行管理
我们认为文件名是不变的,但是储存BED文件路径的向量则可以被命名得更详细。通过makeErmaSet函数可以获取元数据,如下所示:
|
|
使用表格描述染色质状态信息
定义染色体状态与标记概述
下表显示了不同类型的染色质标记是如何组合并用于判断染色体质片段的状态的。那些缩写的意义为:活跃TSS(TssA),两种类型的上下流启动子(PromU, PromD1, PromD2),转录为5’或3‘优先或略强或弱(Tx5’, Tx3’, Tx, TxWk),联合转录或调控(TxReg),优先转录,增强子活性和可能弱(TxEnh5‘,TxEnh3‘,TxEnhW),两种类型的活跃增强子,可能是侧翼或弱的(EnhA1,EnhA2,EnhAF,EnhW1,EnhW2)或由H3K27ac(EnhAc),初级DNase(DNase),ZNF基因和重复序列(ZNF/rpts),异染色质(Het),二价或平衡型启动子(Propp,PromBiv),抑制多梳(ReprPC)或静默(Quies)。
|
|
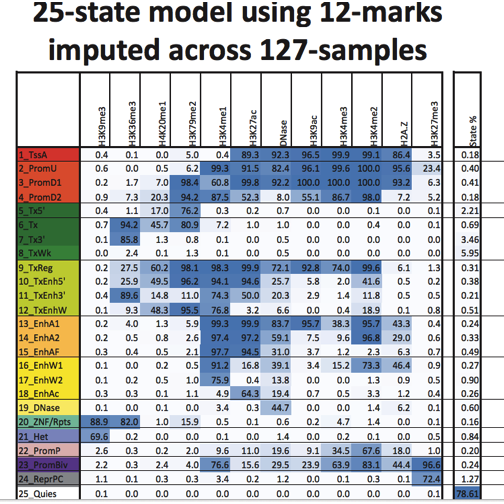
erma包中含有的文件包括31个不同细胞系和基因组的信息(原文里面是tilings,这个我的理解就是表示了一些信息),并根据上面显示了状态图将这些信息分配给染色体状态。
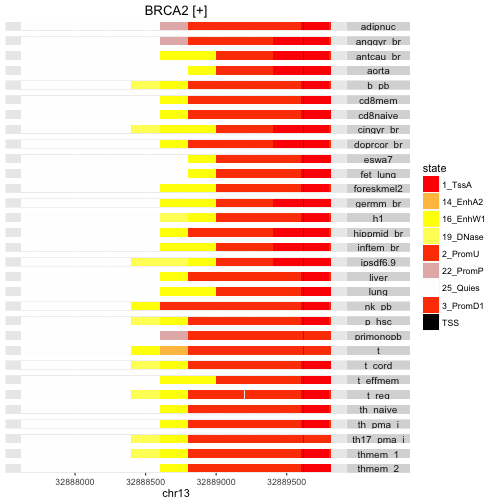
下面展示了两个基因BRCA2和Eome启动子区域的状态标记,如下所示:
|
|
|
|
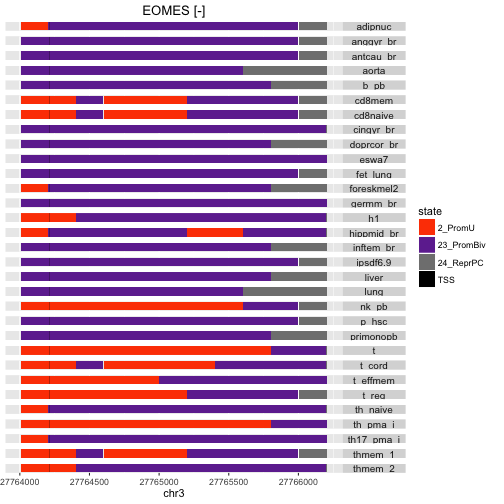
有两个基因在启动子区域的状态与常规的状态不同,以及与细胞类型的一致性方面也不同。
并行目标查询
虽然可视化很直观,但是我们也希望能够进行文本编辑,从而提取一些信息供下游使用。在这里我们简单地将每个细胞类型的状态制成表格。使用readuceByFile函数就能将文件分配给可用的内核或主机进行并行处理,如下所示:
|
|
因此,我们看到,在某个特定的基因的启动子区域的基因序列方面,不同的细胞类型存在着数量和状态类型的变化。在后面的练习中,我们将会考虑如何以这种方式开发其它潜在感兴趣的信息。
总结
在这一部分中我们看到:
- 在磁盘中,使用了大量添加了索引的文本来表示基因组注释或实验结果(原文是:“leave on disk” large numbers of indexed textual representations of genomic annotation or experimental result);
- 使用R包的方式来简化标记这些数据;
- 使用
GenomicFiles来管理有关文件的元数据; - 使用
reduceByFile来并行处理文件内容。
这些技术避免了创建,加载大量的R对象,从而提供了某种程度的便捷性。在内存资源有限的情况下,对于那些需要大量集群(clusters)的应用来说,这种策略就显得性价比较高。